
Foreordained or predestined: complex theological debates have raged around these terms for centuries. Finally science has weighed in, and the correct answer appears to “preordained.” Maybe not for the human soul, but at least for the building blocks of mortality, our amino acids. A Sept. 10 news item from the science website, Phys.org, has this intriguing headline: “Scientists find biology’s optimal ‘molecular alphabet’ may be preordained.” Here’s the story:
An international and interdisciplinary team working at the Earth-Life Science Institute (ELSI) at the Tokyo Institute of Technology has modeled the evolution of one of biology’s most fundamental sets of building blocks and found that it may have special properties that helped bootstrap itself into its modern form.
All life on Earth uses an almost universal set of 20 coded amino acids (CAAs) to construct proteins. This set was likely “canonicalized” or standardized during early evolution; before this, smaller amino acid sets were gradually expanded as organisms developed new synthetic proofreading and coding abilities. The new study, led by Melissa Ilardo, now at the University of Utah, explored how this set evolution might have occurred.
There are millions of possible types of amino acids that could be found on Earth or elsewhere in the universe, each with its own distinctive chemical properties. Indeed, scientists have found these unique chemical properties are what give biological proteins, the large molecules that do much of life’s catalysis, their own unique capabilities. The team had previously measured how the CAA set compares to random sets of amino acids and found that only about 1 in a billion random sets had chemical properties as unusually distributed as those of the CAAs.
The team thus set out to ask the question of what earlier, smaller coded sets might have been like in terms of their chemical properties. There are many possible subsets of the modern CAAs or other presently uncoded amino acids that could have comprised the earlier sets. The team calculated the possible ways of making a set of 3-20 amino acids using a special library of 1913 structurally diverse “virtual” amino acids they computed and found there are 1048 ways of making sets of 20 amino acids.
In contrast, there are only ~ 1019 grains of sand on Earth, and only ~ 1024 stars in the entire universe. “There are just so many possible amino acids, and so many ways to make combinations of them, a computational approach was the only comprehensive way to address this question,” says team member Jim Cleaves of ELSI. “Efficient implementations of algorithms based on appropriate mathematical models allow us to handle even astronomically huge combinatorial spaces,” adds co-author Markus Meringer of the German Center for Air and Space.
As this number is so large that they used statistical methods to compare the adaptive value of the combined physicochemical properties of the modern CAA set with those of billions of random sets of three to 20 amino acids. What they found was that the CAAs may have been selectively conserved during evolution due to their unique adaptive chemical properties, which help them to make optimal proteins, in turn helping organisms that could produce those proteins become more fit.
They found that even hypothetical sets containing only one or a few modern CAAs were especially adaptive. It was difficult to find sets even among a multitude of alternatives that have the unique chemical properties of the modern CAA set. These results suggest that each time a modern CAA was discovered and embedded in biology’s toolkit during evolution, it provided an adaptive value unusual among a huge number of alternatives, and each selective step may have helped bootstrap the developing set to include still more CAAs, ultimately leading to the modern set.
If true, the researchers speculate, it might mean that even given a large variety of starting points for developing coded amino acid sets, biology might end up converging on a similar set. As this model was based on the invariant physical and chemical properties of the amino acids themselves, this could mean that even life beyond Earth might be very similar to modern Earth life. Co-author Rudrarup Bose, now of the Max Planck Institute of Molecular Cell Biology and Genetics in Dresden, further hypothesizes, saying “Life may not be just a set of accidental events. Rather, there may be some universal laws governing the evolution of life.”
In other words, our set of 20 coded amino acids, the key toolkit of life, displays uncanny suitability for making complex proteins. They are so wonderfully tuned that we might even be able to expect life elsewhere in the universe to be be using pretty much the same set. As Dr. Rudrarup Bose put it, “Life may not be just a set of accidental events.” Our amazing molecular machinery appears to have been beautifully endowed with ideal properties by “some universal laws” or something.
The study referred to by Phys.org is Melissa Ilardo et al., “Adaptive Properties of the Genetically Encoded Amino Acid Alphabet Are Inherited from Its Subsets,” Scientific Reports, volume 9, article number 12468 (28 Aug. 2019). The authors focus on only 3 of the most important (among many) properties of amino acids related to solubility, acidity (pKa, the logarithmic acid dissociation constant), and size. These parameters are viewed as part of a chemical space (think of a 3D chart with axes for each parameter). The goal for a useful amino acid toolkit would be to cover a broad space “smoothly” without heavy clustering. Amino acids that are well dispersed in that 3D space would appear to be more capable of achieving the diverse needs of proteins than ones that gives less ideal distribution in chemical property space. Here is an excerpt regarding the approach:
To describe the properties of the molecules in the computed sets, we used the same descriptors previously reported: van der Waals volume (Vvdw), logarithmic acid dissociation constant (pKa, considered specifically over the range from 2–14 here) and partition coefficient (logP), which were selected based on their ability to characterize the functional chemistry space of α-amino acids (cf. refs3,37). Vvdw is simply a measure of the volume of space occupied by the amino acid, which is expected to play a role in mediating steric interactions. LogP describes the affinity of a molecule to a hydrophilic or hydrophobic solvent. In the context of protein structure, this is an important factor in protein folding, and is essential for the heterogeneous nature of protein surface potentials, which is essential for catalysis. pKa describes the pH at the mid-point of a proton transfer by the amino acid side chain, which influences the charged state of an amino acid residue. This in turn affects a host of interactions within and among proteins and their substrates.
While thousands of additional descriptors exist38, we selected what we considered to be fundamental properties that define molecular interactions. This selection was made to minimize bias introduced by considering instead the properties through which we functionally characterize amino acids in modern biochemical contexts. Additionally, these properties are reliably predicted and quantified through chemical property prediction software, an important consideration given the theoretical and computational nature of our data set. This analysis is exploratory, requiring broad, and simplified, choices of molecular descriptors. A previous publication20 offers a specific investigation that justifies the descriptors relating to size, pKa and hydrophobicity, particularly in the light of older work39 that has been built on productively40,41. It is striking that such simple metrics are able to classify amino acid chemical space so effectively2,3.
We used the definition of optimality that was previously introduced to test the CAA alphabet, as described in Ilardo et al.3, namely that more “optimal” sets are those that have broader range and/or evenness of coverage of chemical space. “Better” or “more optimal” sets are those that cover chemical space in the three descriptor categories (Vvdw, pKa and logP) both more broadly and evenly than a comparison set.
Some Background Thoughts
One of the many mysteries of life on earth is the way DNA encodes directions to create the many complex proteins that create and maintain our bodies. All the mind-boggling complexity of proteins and the bizarre machines they create are achieved using just 20 different building blocks called amino acids. A simple coding system in our DNA relies on different combinations of just four bases, adenine (A), guanine (G), cytosine (C), and thiamine (T), to represent any of the 20 amino acids by a “codon” made from a sequence of three bases. Long sequences of these codons encode the genes, proteins, regulators, etc. in our DNA, and the resulting series of amino acids joined together when proteins are made from our genes create amazing tools that carry out numerous essential functions. The coding of information with four bases to define a series of amino acids seems so simple but gives such incredible complexity in the end. The way a series of amino acids can become twisted, wrapped, and folded layer upon layer into bizarre 3-dimensional shapes is a topic for wonder and much further research, but it all depends on the properties of our 20 basic building blocks. But why those 20 amino acids? Here’s a table showing the chemical structure of these 20 coded amino acids from Particle Sciences.
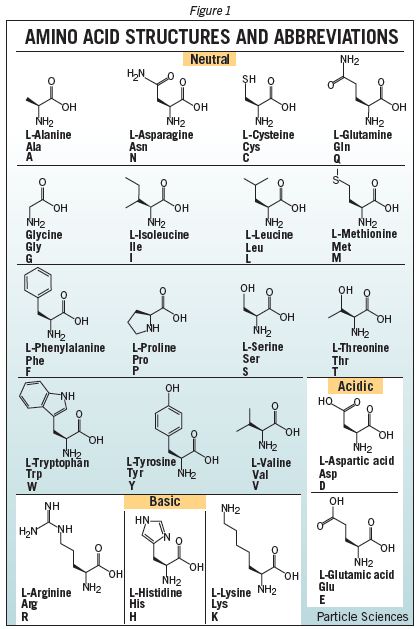
Looking at the list of amino acids, it’s easy for pundits to see some slop and inefficiency. Some of the 20 amino acids are very similar. It’s easy to think that a smaller set could be used that could make our DNA much more efficient. It’s easy to think that the apparent inefficiency is what you can expect when random choices are made in nature that lead to something that works that a hypothetical intelligent designer could have made much more efficient. An example is provided by Stuart Cantril, “They’d none of them be missed – why 20 amino acids and not 15?,” a post on a leading chemistry blog at Nature.com, The Skeptical Chymist, from 2008. Cantril suggests that we don’t really need all 20 amino acids and would have a far more efficient set if nature had been smart enough to just use 15. I don’t know if the new study changes any of the speculations raised there, but if indeed all 20 amino acids appear to be optimized and cannot readily be improved by swapping them out for others, then it would seem that the subtle differences between some of them must play important functional roles, perhaps like a Phillips head versus a slotted (flat head) screwdriver. A good toolkit needs both (though a screwdriver with many detachable heads is the more intelligent choice).
Cantril argues, for instance, that several amino acids with hydrocarbon side chains of different length seem redundant. “Do we really need 4 different hydrocarbon side chains (methyl, isopropyl, sec-butyl, isobutyl)? … Get rid of two of them – probably a long one and a short one.” This may have been answered adequately by Andrew J. Doig in “Frozen, but no accident – why the 20 standard amino acids were selected,” FEBS Journal, Volume 284, Issue 9 (May 2017): 1296-1305. Doig discusses the four amino acids with hydrocarbon side chains, Val, Leu, Ile, Phe (valine, leucine, isoleucine and phenylanaline) and explains why they are needed in spite of being similar:
These hydrocarbon side chains are clearly present to drive protein folding by forming hydrophobic cores, yet why exactly these were selected deserves an explanation. Firstly, why are there so many hydrocarbon side chains, rather than just, say, Leu? Multiple hydrocarbon side chains may be used as they are required to fill a protein core with no clashes and no holes. A variety of pieces are required to fit all the gaps within a protein core. Each can adopt a number of rotamers [variants of a molecule that differ in how a portion of the molecule has been rotated] with similar energies, thus giving a range of possible shapes for each side chain. Thus, Leu and Ile are both needed to increase the range of possible hydrocarbon 3D shapes, despite Ile being more costly to synthesise….
Val, Leu, Ile and Phe are striking in that they all have branched carbons, rather than straight chains. Using a branch gives one fewer dihedral angle to be fixed compared to a straight chain. Side chains therefore enter a protein core not just because they are hydrophobic, but also because they do not lose too much conformational entropy when they fold. Similarly, rings are rigid, so Phe has a large hydrocarbon surface and only two bonds to be fixed.
Hydrocarbon side chains larger than Phe, Ile or Leu may not be used, as they would be less soluble as amino acids. A cyclohexane ring is also less soluble than a benzene ring, so Phe is used, rather than cyclohexylalanine.
The point is that achieving the desired 3D shape of the protein and having it be stable, not “mush” that changes shape unpredictably, is vital for protein function, and these related amino acids help give the 3D shape needed of the units within the folded protein to fill up the space inside and do that efficiently without losing other properties such as solubility.
On the other hand, Doig is not impressed with methionine. He argues that this sulfur-containing amino acid might have been more useful back when the earth lacked an oxygen-rich atmosphere, but now is a non-ideal amino acid that we are “stuck” with. But in light of Melissa Ilardo et al.’s publication, it would seem to be a unique structure gives some significant benefits that can’t be obtained without it. Of methionine specifically, the authors write:
Conversely, Met [methionine] appears to be advantageous in small set sizes compared to the possibilities within the XAAs [xeno amino acids, the set of alternate amino acids considered]. However, it is not until larger set sizes that Met begins to become adaptive when looking at the coded set. This could indicate that Met does not offer an enormous adaptive advantage to the encoded set as a whole, however, there are very few possibilities within the XAA library that have similar chemical properties to Met, and therefore Met is uniquely advantageous to encoded sets.
The authors also point how how leucine and isoleucine play an important cooperative role in their extensive numerical analysis, making it appropriate that the code amino acids would include both, in spite of the tendency of chemists to see them as nearly the same.
Fascinating work, intriguing implications. Thanks be to “universal laws.”
Update, Oct. 1, 2019
Andrew Doig’s commentary on the role and selection of cysteine (Cys) as a coded amino acid is also noteworthy, especially the second paragraph below:
A key function of proteins is to bind metals. Soft metals, such as copper, zinc and cadmium, bind more tightly to sulfur than to oxygen. Cys may therefore have been selected for its metal binding properties, despite its high biosynthetic cost (Table 1). In particular, Cys is commonly used to bind iron‐sulfur clusters. These cofactors are found in a wide variety of metalloproteins, are playing crucial roles in metabolism and are very ancient [43]. The SH side chain is also an effective nucleophile; it has a lower pKa than OH, readily forming S−.
Cys is the only side chain able to perform redox reactions, by forming disulfide bonds to stabilise folded proteins. Cys was selected in an anaerobic environment, however, where disulfide bond formation would have been rare or nonexistent. It is therefore not plausible that Cys was selected for its ability to form disulfides, and its subsequent use for this purpose, starting very approximately 1.5 billion years later after the Great Oxidation Event, is a lucky accident.
Lucky accident, or “preordained” wisdom from a wise universal law or something? Every aspect of life itself, from the miracle of efficient carbon synthesis in stars, the balance between electromagnetic and gravitational forces that allow stars to exist perched on the razor’s edge between violent explosion on one hand and gravitational collapse into oblivion on the other, the many wonders of water, the architecture of Earth’s core and its protective magnetic field, and the many miracles of amino acids and proteins (not the least of which are the bizarre and intricately organized “lucky beyond imagination” machinery of FTP synthase and the spliceosome), all calling attention to lucky accident after lucky accident in ways that suggest that our cosmos and this planet has been rigged for life.
To me, it’s beyond the question of “could this have happened by chance?” The real question, after pondering something like the splicesome that turns our 20,000 genes into over 100,000 useful proteins by cutting and pasting our encoded amino acid sequences into new beneficial and even essential proteins, is this: “How are such incredible designs, such mind-boggling gadgets and solutions, even possible?”
That a universe tuned to allow stars to exist could also be tuned to make stars efficient carbon creators is amazing. That it could also give us atoms that could provide us with a nearly universal solvent so vital for life that has the incredibly rare, even bizarre feature of expanding when it freezes — so that sinking ice doesn’t fill the oceans from the bottom up but stays on top to insulate them — is fortunate indeed. And after all that, to have amino acids that could not only be so efficient to support all the intricacies of encoding proteins and ensuring that the proteins fold and function properly in their aqueous environment, but could also give us a protein-based cut-and-paste machine (the spliceosome, as discussed here previously) that mixes and matches amino acids from our genes to allow many more proteins to be encoded beyond what are fit into the DNA in the tiny nucleus of the cell, and do this in a way that doesn’t destroy needed proteins or create random lethal junk, well, it’s just getting lucky beyond belief, or rather, lucky in a way that should strengthen belief for those open to the possibility of the existence of a Creator.
Related reading:
- “Protein Structure,” from Particle Sciences. 2009. Describes the basics of protein structure, including primary, secondary, teriary, and quaternary structure, with some hints about how the different amino acids affect the highly complex and intricate structure that a protein chain can end up having after assembly.









Fascinating, though I see nothing in it to support intelligent design or theistic evolution.
— OK
Fascinating
Perhaps "intelligent direction" would be a better term to use. It looks quite unlikely at this point, on the basis of this kind of study and others of its kind, that complex life on earth would have come about without things having been directed intelligently.
But, why is there any matter anyway? Why not nothing?
OK said, "Fascinating, though I see nothing in it to support intelligent design or theistic evolution."
I especially feel that way myself when I look at one of the many random downstream byproducts of amino acid evolution, Microsoft Windows. Still, there are folks who sincerely insist that it has some evidence of intelligent design. Believers gotta believe.
… and Microsoft has a trillion dollar valuation. If that fact doesn't convince you there is no God, nothing will.
My apologies to Microsoft. I actually like many aspects of Microsoft products. But I have had some faith-shaking episodes with a number of updates that seemed more like the result of random mutations than planned progress.
How quickly he bows before the golden calf.